Research Topics
Integration of Machine Learning and High-Performance Computing
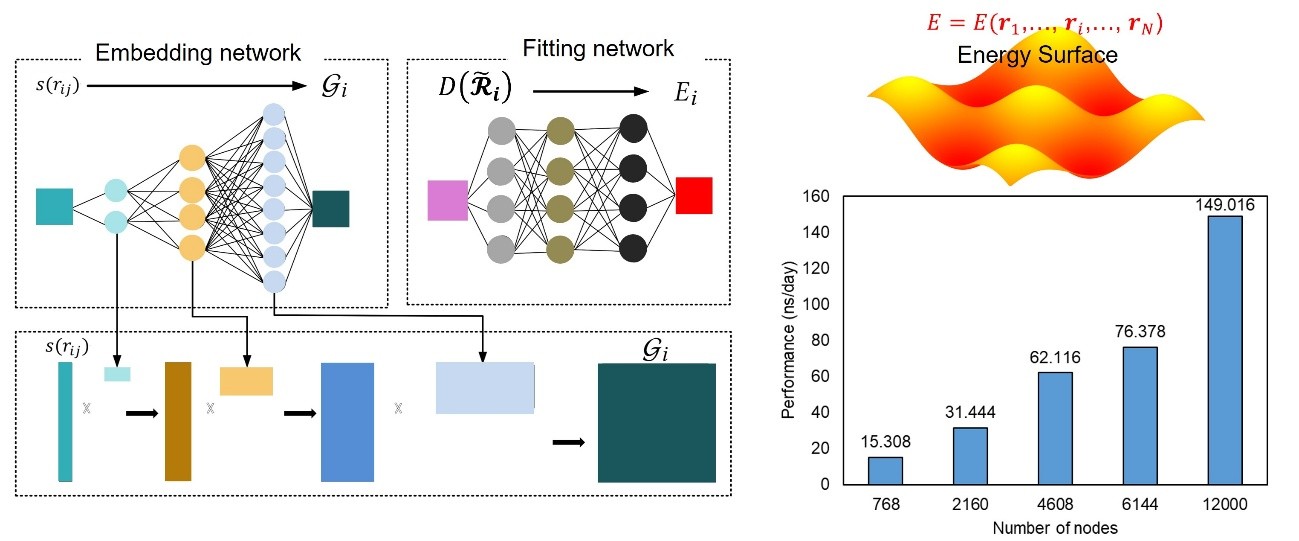
The optimization of material structures and the design of new materials rely on accurate property predictions through high-precision computational methods. While conventional macroscale modeling addresses real-scale simulations, it often depends on empirical approaches that limit accuracy. Conversely, atomistic-level techniques like ab initio molecular dynamics (AIMD) offer high precision but come with high computational costs, restricting the time and spatial scales that can be simulated. As chemical reactions and phase transitions occur on millisecond to microsecond time scales, reproducing these phenomena with AIMD is challenging, highlighting the need for rapid, high-precision analytical methods. In this study, we employed a machine learning-based interatomic potential model for optimization on the Fugaku supercomputer, maintaining the accuracy of potential energy and interatomic forces. This approach resulted in a 31.7-fold increase in the computational speed of molecular dynamics (MD) simulations, achieving 149 nanoseconds per day across 12,000 nodes while retaining ab initio calculation accuracy.
Integration of Machine Learning and High-Performance Computing
The optimization of material structures and the design of new materials rely on accurate property predictions through high-precision computational methods. While conventional macroscale modeling addresses real-scale simulations, it often depends on empirical approaches that limit accuracy. Conversely, atomistic-level techniques like ab initio molecular dynamics (AIMD) offer high precision but come with high computational costs, restricting the time and spatial scales that can be simulated. As chemical reactions and phase transitions occur on millisecond to microsecond time scales, reproducing these phenomena with AIMD is challenging, highlighting the need for rapid, high-precision analytical methods. In this study, we employed a machine learning-based interatomic potential model for optimization on the Fugaku supercomputer, maintaining the accuracy of potential energy and interatomic forces. This approach resulted in a 31.7-fold increase in the computational speed of molecular dynamics (MD) simulations, achieving 149 nanoseconds per day across 12,000 nodes while retaining ab initio calculation accuracy.
Evaluation of Mechanical Properties of Magnesium Alloys
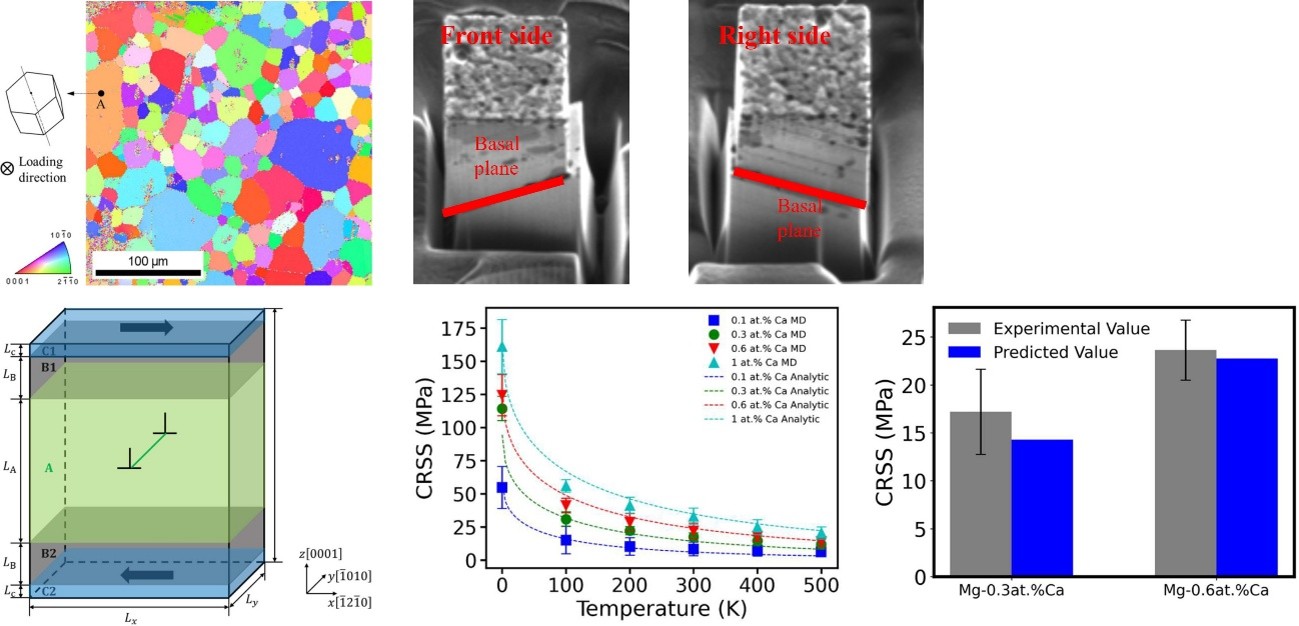
Magnesium (Mg) is recognized as the lightest structural metal and serves as a promising alternative to heavier steels and aluminum alloys. Nevertheless, Mg exhibits limited plastic formability at room temperature and generally displays inferior strength compared to most aluminum alloys. This constraint in formability can be attributed to the significant disparity in critical resolved shear stress (CRSS) between basal slip systems and other slip and twinning systems. Consequently, the low CRSS acts as a limiting factor for the strength of Mg alloys, underscoring the importance of enhancing basal slip to improve their strength, deformability, and fracture toughness. This study presents a theoretical model aimed at predicting the CRSS in single-crystal Mg–Ca alloys. We quantitatively investigated the CRSS of basal dislocation slip along the basal plane using molecular dynamics simulations across a temperature range of 1 K to 500 K. The results obtained from the proposed theoretical model agree well with both simulation findings and micro-pillar compression test results for Mg–0.3 at.% Ca and Mg–0.6 at.% Ca alloys at ambient temperature.
Evaluation of Mechanical Properties of Magnesium Alloys
Magnesium (Mg) is recognized as the lightest structural metal and serves as a promising alternative to heavier steels and aluminum alloys. Nevertheless, Mg exhibits limited plastic formability at room temperature and generally displays inferior strength compared to most aluminum alloys. This constraint in formability can be attributed to the significant disparity in critical resolved shear stress (CRSS) between basal slip systems and other slip and twinning systems. Consequently, the low CRSS acts as a limiting factor for the strength of Mg alloys, underscoring the importance of enhancing basal slip to improve their strength, deformability, and fracture toughness. This study presents a theoretical model aimed at predicting the CRSS in single-crystal Mg–Ca alloys. We quantitatively investigated the CRSS of basal dislocation slip along the basal plane using molecular dynamics simulations across a temperature range of 1 K to 500 K. The results obtained from the proposed theoretical model agree well with both simulation findings and micro-pillar compression test results for Mg–0.3 at.% Ca and Mg–0.6 at.% Ca alloys at ambient temperature.
Analysis of Organic Field-Effect Transistors
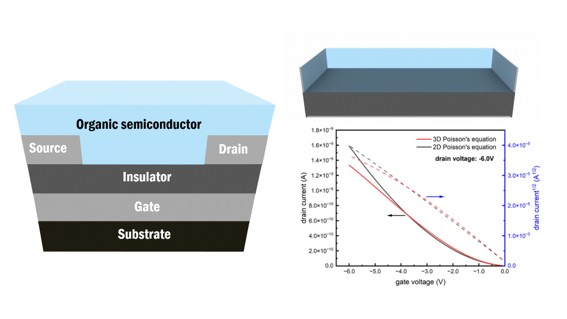
The Organic Field-Effect Transistor (OFET) is recognized as a pivotal device in applications such as flexible circuits, sensors, and memory storage. However, its performance typically lags behind that of inorganic counterparts. Enhancing the performance of OFETs necessitates precise control over the characteristics of the organic semiconductor layer. Recent advancements in molecular-level simulation techniques, including kinetic Monte Carlo methods, have significantly enhanced the capability to replicate the microscopic behavior of charge carriers. Nevertheless, these approaches often depend on two-dimensional Poisson equations, which inadequately capture short-range electrostatic interactions. In this research, we successfully employed a three-dimensional partial differential equation solver to model OFET devices, fully accounting for the Coulomb interactions between charge carriers. Our findings reveal that at elevated gate voltages, the transfer characteristics derived from the three-dimensional Poisson equation exhibit a downward curvature and lower values compared to those predicted by two-dimensional Poisson equations, suggesting strong concordance with experimental observations.
Analysis of Organic Field-Effect Transistors
The Organic Field-Effect Transistor (OFET) is recognized as a pivotal device in applications such as flexible circuits, sensors, and memory storage. However, its performance typically lags behind that of inorganic counterparts. Enhancing the performance of OFETs necessitates precise control over the characteristics of the organic semiconductor layer. Recent advancements in molecular-level simulation techniques, including kinetic Monte Carlo methods, have significantly enhanced the capability to replicate the microscopic behavior of charge carriers. Nevertheless, these approaches often depend on two-dimensional Poisson equations, which inadequately capture short-range electrostatic interactions. In this research, we successfully employed a three-dimensional partial differential equation solver to model OFET devices, fully accounting for the Coulomb interactions between charge carriers. Our findings reveal that at elevated gate voltages, the transfer characteristics derived from the three-dimensional Poisson equation exhibit a downward curvature and lower values compared to those predicted by two-dimensional Poisson equations, suggesting strong concordance with experimental observations.



© L. LIU GROUP, 2026
Contact Information for Lijun Liu
E-mail : liu@mech.eng.osaka-u.ac.jp
Access : Department of Mechanical Engineering, The University of Osaka, Bldg. No. M1, Room 726, 2-1 Yamadaoka, Suita 565-0871, Japan
Contact Information for Lijun Liu
E-mail : liu@mech.eng.osaka-u.ac.jp
Access : Department of Mechanical Engineering, The University of Osaka, Bldg. No. M1, Room 726, 2-1 Yamadaoka, Suita 565-0871, Japan
© L. LIU GROUP, 2026